درباره بیماری و روشهای تشخیص
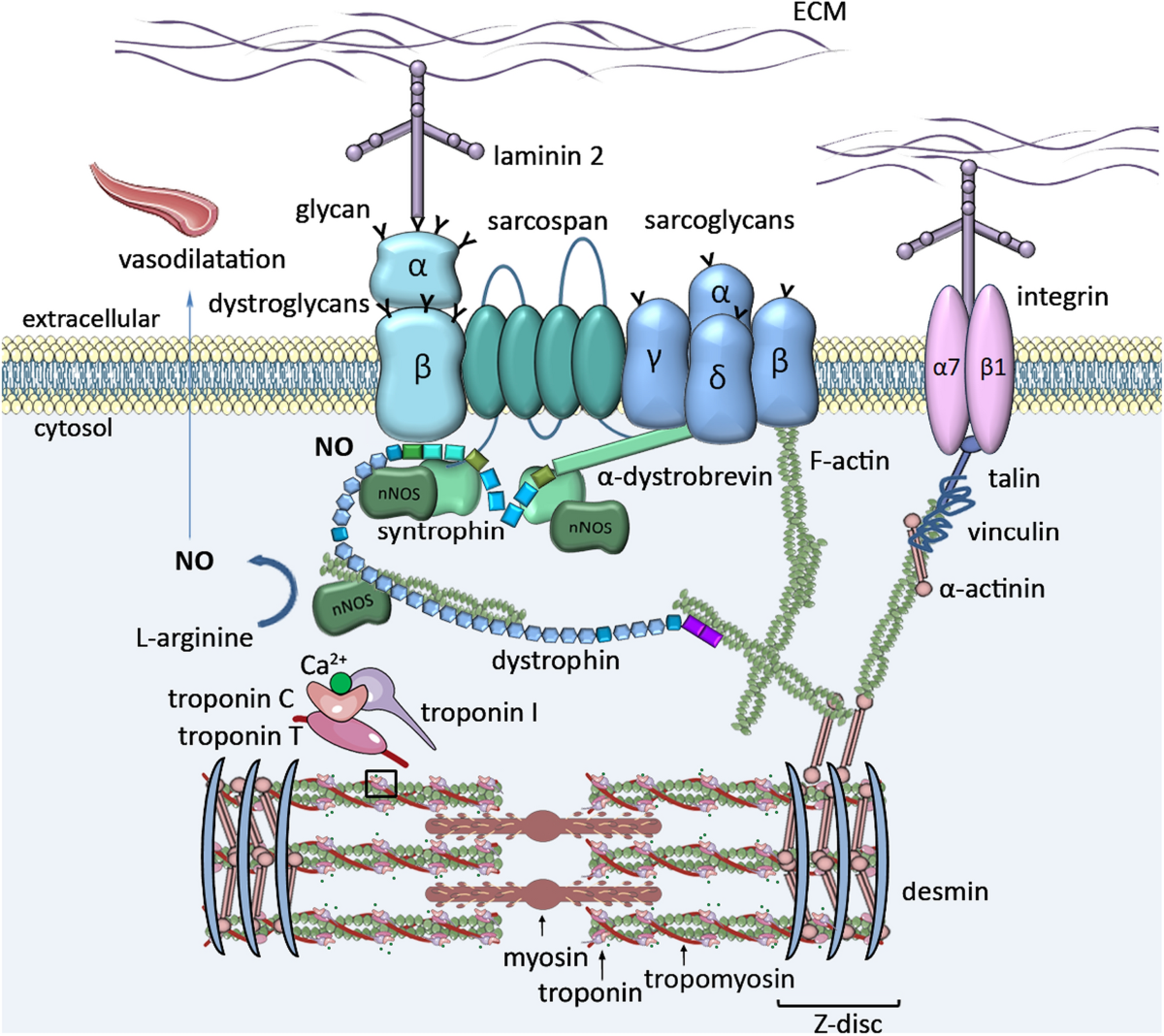
دیستروفی عضلانی دوشن (DMD) و بکر (BMD) از جمله شایعترین بیماریهای ارثی عضلانی هستند که در اثر جهش در ژن DMD، واقع در کروموزوم X، ایجاد میشوند. این ژن مسئول تولید پروتئینی به نام دیستروفین است که نقش حیاتی در حفظ ساختار و عملکرد صحیح سلولهای عضلانی دارد.
تفاوت دوشن و بکر
- دوشن (DMD): فرم شدید بیماری که علایم آن از دوران کودکی (معمولاً قبل از سن ۵ سالگی) آغاز میشود و به سرعت پیشرفت میکند. بیشتر مبتلایان تا سن نوجوانی توانایی راه رفتن را از دست میدهند.
- بِکِر (BMD): فرم خفیفتر که شروع علایم آن دیرتر (معمولاً در سنین نوجوانی یا بزرگسالی) است و سیر پیشرفت آن کندتر میباشد. این بیماران ممکن است تا دهه سوم زندگی توانایی راه رفتن را حفظ کنند.
علایم و نشانههای بالینی
- ضعف پیشرونده عضلات لگن، ران و شانه
- مشکل در برخاستن از زمین (علامت گاور)
- افتادنهای مکرر
- بزرگی عضلات ساق پا بهدلیل فیبروز (Pseudohypertrophy)
- مشکلات قلبی (کاردیومیوپاتی)
- مشکلات تنفسی در مراحل پیشرفته
- در برخی موارد، اختلالات یادگیری و توجه نیز مشاهده میشود بهویژه در (DMD)
الگوی وراثت
این بیماری با الگوی وابسته به X مغلوب منتقل میشود. بنابراین بیشتر مبتلایان پسر هستند و زنان ناقل بدون علایم یا با علایم خفیف میباشند. هر مادر ناقل، در هر بارداری پسر، ۵۰٪ احتمال دارد فرزندی مبتلا داشته باشد.
روشهای تشخیص
- آنزیم کراتین کیناز (CK): به شدت افزایش یافته و نشانه آسیب عضلات است.
- الکترو میوگرافی (EMG): نشاندهنده ماهیت میوپاتیک آسیب عضلات.
- بیوپسی عضله:کاهش یا عدم وجود دیستروفین را نشان میدهد.
- آزمایش ژنتیک: بررسی جهشهای حذف یا اضافه در ژن DMD با روشهای MLPA یا NGS
- بررسی حاملگی در زنان ناقل: با آزمایش ژنتیکی روی مادر و بررسی جنین از طریق آمنیوسنتز یا CVS
نوع نمونه
- خون محیطی
- نمونه بافت عضله در موارد خاص برای ایمونوهیستوشیمی
- مایع آمنیوتیک برای تشخیص پیش از تولد
مقدار نمونه
- خون محیطی: ۳ تا ۵ میلیلیتر
- نمونه بافت: حدود ۵۰ میلیگرم
- مایع آمنیوتیک: 20 میلیلیتر
شرایط نگهداری نمونه
- خون: در دمای یخچال (۲ تا ۸ درجه سانتیگراد) تا ۷۲ ساعت
- نمونه بافتی: در سرم فیزیولوژی و در دمای یخچال، برای انتقال سریع
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- تست CK: ۱ تا ۲ روز کاری
- آزمایش ژنتیک (NGS): ۴ تا ۶ هفته
- بررسی ژنتیکی پیش از تولد: ۴ تا ۶ هفته
منابع علمی
- Bushby, K., et al. (2010). “Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management.” The Lancet Neurology, 9(1), 77–93.
- Flanigan, K. M. (2014). “Duchenne and Becker muscular dystrophies.” Neurologic Clinics, 32(3), 671–688.
- Hoffman, E. P., Brown, R. H., & Kunkel, L. M. (1987). “Dystrophin: the protein product of the Duchenne muscular dystrophy locus.” Cell, 51(6), 919–928.
- Aartsma-Rus, A., & Ginjaar, I. B. (2016). “Duchenne and Becker muscular dystrophy: The same but different.” Neuromuscular Disorders, 26(8), 447–448.
- Mah, J. K., et al. (2017). “A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy.” Neuromuscular Disorders, 27(1), 3–15.
")
")