درباره بیماری و روشهای تشخیص
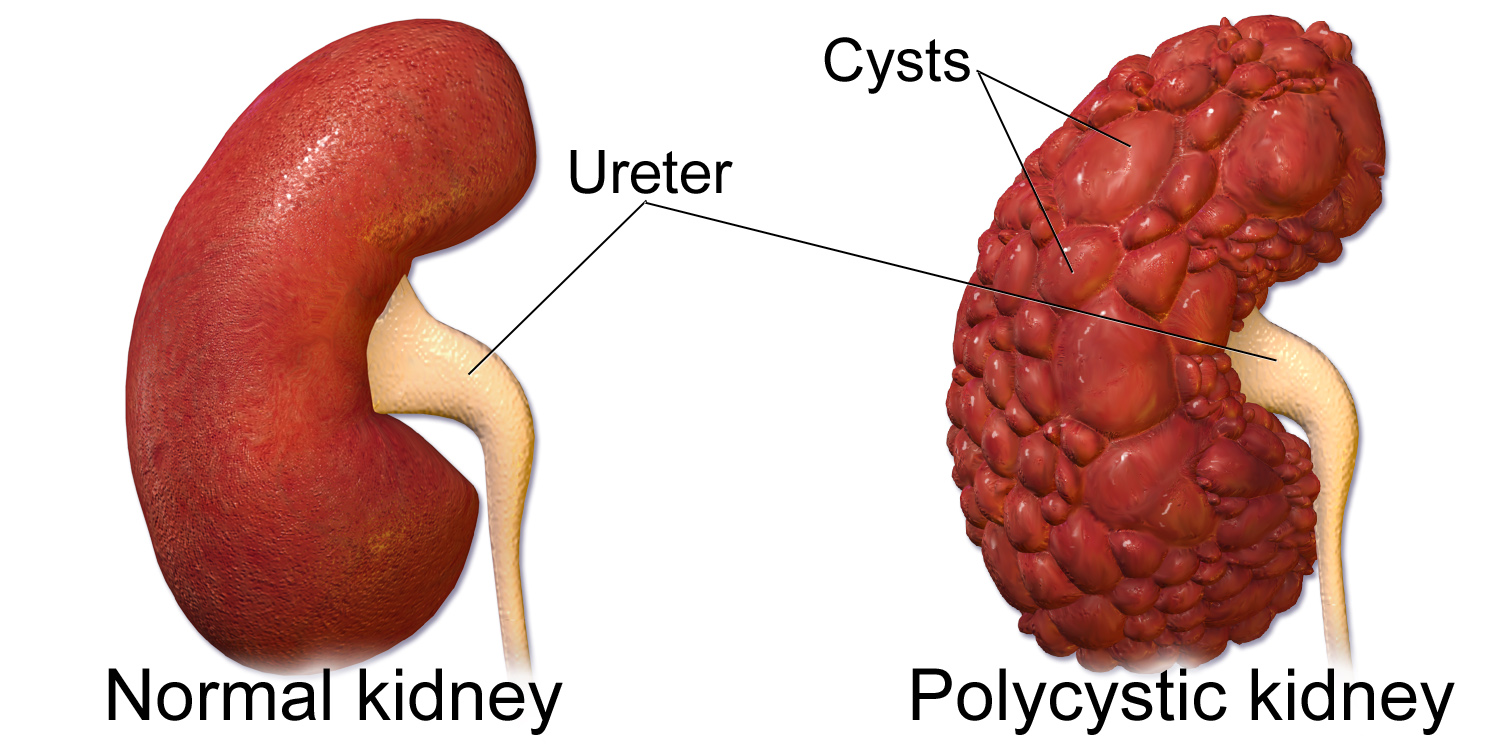
بیماری کلیه پلیکیستیک نوعی بیماری ژنتیکی است که با تشکیل کیستهای متعدد پر از مایع در کلیهها مشخص میشود. این کیستها بهتدریج در طول زمان رشد کرده و عملکرد طبیعی کلیه را مختل میکنند. این بیماری میتواند به نارسایی کلیه منجر شود و یکی از شایعترین دلایل نارسایی کلیوی ارثی در جهان است.
انواع بیماری
بیماری کلیه پلیکیستیک دو نوع اصلی دارد:
- نوع اتوزوم غالب (ADPKD) :
شایعترین نوع این بیماری است که معمولاً در دهه سوم یا چهارم زندگی تظاهر مییابد. تقریباً از هر 1000 نفر، 1 نفر به آن مبتلا میشود. این نوع معمولاً ناشی از جهش در ژنهای PKD1 (حدود 85% موارد) و PKD2 (حدود 15% موارد) است.
- نوع اتوزوم مغلوب (ARPKD) :
این نوع نادرتر و شدیدتر بوده و معمولاً در دوران نوزادی یا کودکی بروز میکند. عامل ژنتیکی اصلی آن جهش در ژن PKHD1 است که روی کروموزوم 6 قرار دارد.
علایم و نشانههای بالینی
علایم در نوع غالب معمولاً در بزرگسالی ظاهر میشود و شامل موارد زیر است:
- درد مزمن در پهلو یا شکم
- فشار خون بالا
- هماچوری (وجود خون در ادرار)
- عفونتهای مکرر دستگاه ادراری
- سنگ کلیه
- نارسایی تدریجی کلیه
- اتساع شکم و لمس تودههای کلیوی در برخی موارد
در نوع مغلوب (ARPKD)، علایم ممکن است در زمان تولد بروز یابد:
- بزرگی شکم به دلیل کلیههای متورم
- نارسایی تنفسی در بدو تولد
- نارسایی کبدی
- فشار خون بالا از کودکی
- اختلال در رشد کودک
روشهای تشخیص
تشخیص PKD با تکیه بر تصویربرداری و بررسیهای ژنتیکی انجام میشود:
- سونوگرافی کلیهها: روش ابتدایی و غیرتهاجمی برای تشخیص کیستها
- MRI یاCT-Scan : در صورت نیاز به بررسی دقیقتر
- آزمایش ژنتیکی: برای شناسایی جهش در ژنهای PKD1، PKD2 یا PKHD1، بهویژه در موارد خانوادگی یا تشخیص پیش از تولد
- سابقه خانوادگی: بسیار حائز اهمیت است و باید همواره در ارزیابی بالینی در نظر گرفته شود.
نوع نمونه
- خون محیطی برای انجام آزمایشات ژنتیک
- مایع آمنیوتیک: در تشخیص پیش از تولد
مقدار نمونه
- خون محیطی: ۳ تا ۵ میلیلیتر
- مایع آمنیوتیک: ۱۰ تا ۱۵ میلیلیتر
شرایط نگهداری نمونه
- خون محیطی: در دمای ۲ تا ۸ درجه سانتیگراد (تا ۷۲ ساعت)
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- تستهای ژنتیکی مبتنی بر NGS یا Sanger برای ژنهای PKD1 و PKD2: ۱۴ تا ۲۱ روز کاری
- تشخیص پیش از تولد (PND): معمولاً بین ۷ تا ۱۰ روز کاری
- غربالگری تصویربرداری در صورت نیاز به MRI یا سونوگرافی: در همان روز یا حداکثر ۲ روز
منابع علمی
- Torres, V. E., & Harris, P. C. (2009). “Polycystic kidney disease in adults.” The Lancet, 373(9667), 1287–1301.
- Bergmann, C. (2017). “ARPKD: Challenges, progress and perspectives.” Pediatric Nephrology, 32(4), 479–487.
- Harris, P. C., & Torres, V. E. (2014). “Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease.” Journal of Clinical Investigation, 124(6), 2315–2324.
- Ong, A. C. M., & Harris, P. C. (2015). “A polycystin-centric view of cyst formation and disease: the polycystins revisited.” Kidney International, 88(4), 699–710.
- Bergmann, C., et al. (2005). “Mutations in the PKHD1 gene in ARPKD patients.” Journal of the American Society of Nephrology, 16(10), 3025–3031.
")
")