درباره بیماری و روشهای تشخیص
بیماری شارکوت ماری توث (CMT) یکی از شایعترین بیماریهای ژنتیکی نوروموسکولار است که موجب آسیب به اعصاب محیطی (پریفرال) میشود. این اعصاب مسئول انتقال پیامها از مغز و نخاع به عضلات و برعکس هستند. این بیماری بهطور پیشرونده باعث ضعف، لاغری عضلانی (آتروفی)، تغییر شکل پاها و کاهش حس در اندامها بهویژه پاها میشود.
نام این بیماری برگرفته از نام سه پزشک فرانسوی و بریتانیایی است که برای اولین بار آن را در اواخر قرن ۱۹ توصیف کردند. شیوع آن حدود ۱ نفر در هر ۲۵۰۰ نفر است و ممکن است در هر سنی شروع شود، ولی معمولاً از دوران کودکی یا نوجوانی آغاز میشود.
انواع بیماری CMT
بیماری CMT انواع مختلفی دارد که بسته به ژن معیوب و الگوی آسیبدیدگی عصبی تقسیمبندی میشوند. سه نوع اصلی آن عبارتند از:
- CMT نوع 1: دمیلینهکننده؛ در این نوع غلاف میلین که اطراف رشتههای عصبی را پوشانده است دچار آسیب میشود. شایعترین نوع، CMT1A است که معمولاً ناشی از دو برابر شدن (duplication) ژنPMP22 در کروموزوم 17 p است.
- CMT نوع 2: آکسونال؛ در این نوع، خود آکسون عصبی آسیب میبیند.
- CMTX: شکل وابسته به کروموزوم X، که مردان را شدیدتر مبتلا میکند و ژن مرتبط با آنGJB1 (کدکننده کانکسین 32) است.
علایم بالینی
- ضعف عضلات پاها، بهویژه در ساق و مچ پا
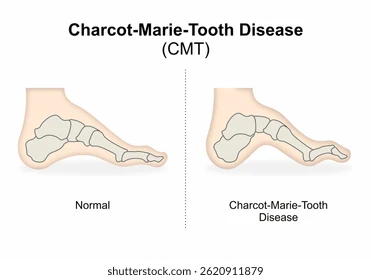
- آتروفی عضلانی (لاغر شدن ساق پا، شکل “پاهای شبیه بطری برعکس”)
- دشواری در راه رفتن، دویدن و حفظ تعادل
- افتادگی پا (foot drop)
- تغییر شکل کف پا ) قوس زیاد یا (flat foot
- کاهش حس لمس، درد یا حرارت
- درگیری دستها در مراحل پیشرفتهتر
علایم معمولاً پیشرونده و غیرکشنده هستند، اما شدت آنها در افراد مختلف متفاوت است. برخی افراد فقط علایم خفیف دارند، در حالی که برخی دیگر به استفاده از وسایل کمکی برای راه رفتن نیاز دارند.
روشهای تشخیص
- معاینه بالینی و سابقه خانوادگی: ارزیابی علایم نورولوژیک و بررسی وجود بیماری در بستگان
- الکترومیوگرافی (EMG) و بررسی هدایت عصبی(NCV): برای تمایز بین نوع دمیلینهکننده و آکسونال
- آزمایش ژنتیک:
- برای شناسایی جهش یا تکرار در ژنهای خاص
- آزمایش MLPA برای بررسی duplications/deletions
- آزمایش NGS که شامل چندین ژن مرتبط با CMT مانند PMP22, MPZ, GJB1, MFN
نوع نمونه
- خون محیطی (EDTA) برای استخراج DNA ژنومی
مقدار نمونه
- ۳ تا ۵ میلیلیتر خون کامل در لولههای حاوی EDTA
شرایط نگهداری نمونه
- نگهداری در دمای یخچال (۲ تا ۸ درجه سانتیگراد)
- نباید منجمد شود
- ارسال نمونه در کمترین زمان ممکن (ترجیحاً ظرف ۲۴ تا ۴۸ ساعت)
مدت زمان جوابدهی
- برای آزمایشهای تکژنی مانند(PMP22) : ۴ تا ۶ هفته
- برای پنل ژنتیکی کاملCMT: ۲ تا ۳ ماه
منابع علمی
- Saporta MA. “Charcot–Marie–Tooth disease and other inherited neuropathies.” Continuum (Minneap Minn). 2014;20(5 Peripheral Nervous System Disorders):1208–1225.
- Pareyson D, Marchesi C. “Diagnosis, natural history, and management of Charcot-Marie-Tooth disease.” Lancet Neurol. 2009;8(7):654–667.
- Rossor AM, Polke JM, Houlden H, Reilly MM. “Clinical implications of genetic advances in Charcot-Marie-Tooth disease.” Nat Rev Neurol. 2013;9(10):562–571.
- Skre H. “Genetic and clinical aspects of Charcot-Marie-Tooth’s disease.” Clin Genet. 1974;6(2):98–118.
- Auer-Grumbach M. “Hereditary sensory neuropathy type I.” Orphanet Journal of Rare Diseases. 2008;3:7.
")
")