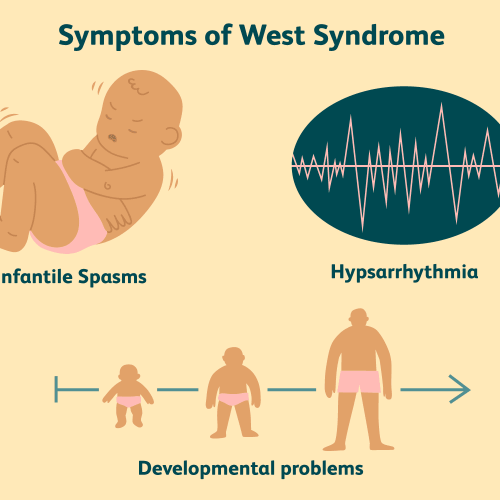
درباره بیماری و روشهای تشخیص
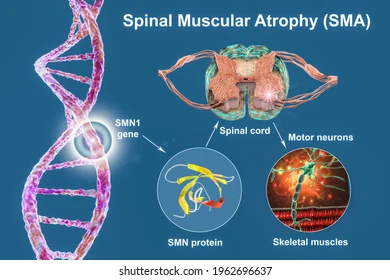
آتروفی عضلانی نخاعی (SMA) یک بیماری ژنتیکی نادر اما جدی است که باعث تحلیل تدریجی سلولهای حرکتی نخاع و در نتیجه ضعف عضلانی، ناتوانی در حرکت و در موارد شدید، مشکلات تنفسی میشود. این بیماری عمدتاً در اثر جهش یا حذف در ژن SMN1 روی کروموزوم ۵ ایجاد میشود. این ژن مسئول تولید پروتئینی به نام SMN (Survival Motor Neuron) است که برای زنده ماندن نورونهای حرکتی حیاتی است.
در نبود پروتئین SMN کافی، نورونهای حرکتی در نخاع از بین میروند و ماهیچههایی که تحت کنترل این نورونها هستند به مرور تحلیل میروند. اگرچه بیماری عمدتاً در کودکان بروز میکند، اما انواع خفیفتر ممکن است تا نوجوانی یا بزرگسالی تشخیص داده نشوند.
انواع SMA
- نوع 1 (وردنیگ-هوفمان): شدیدترین فرم، با شروع در دوران نوزادی (قبل از 6 ماهگی) و امید به زندگی کوتاه، در صورت عدم درمان
- نوع 2: شروع بین 6 تا 18 ماهگی؛ کودک ممکن است بنشیند ولی قادر به راه رفتن نخواهد بود
- نوع 3 (کوگلبرگ-ولاندر): شروع در دوران کودکی یا نوجوانی، با شدت کمتر و توانایی ایستادن یا راه رفتن
- نوع 4: نوع بسیار خفیف با شروع در بزرگسالی، ضعف عضلات آهسته و پیشرونده
علایم بالینی
- ضعف عضلات، بهویژه در پاها و تنه
- کاهش تون عضلانی (هیپوتونی)
- مشکلات در بلع و تغذیه در نوزادان
- ناتوانی در ایستادن یا راه رفتن (بسته به نوع بیماری)
- اختلالات تنفسی (در انواع شدید)
روشهای تشخیص
غربالگری نوزادان
در بسیاری از کشورها، آزمایش ژنتیکی برای SMA بخشی از برنامه غربالگری نوزادان است.
آزمایش ژنتیکی
- بررسی وجود حذف در اگزون 7 از ژن SMN1
- بررسی تعداد کپی ژن SMN2 (ژن پشتیبان) که در شدت بیماری تأثیر دارد
- روشهای مورد استفاده:
- MLPA (Multiplex Ligation-dependent Probe Amplification)
- qPCR
- توالییابی نسل جدید یا NGS در موارد پیچیده یا خانوادههای در معرض خطر
نوع نمونه
- خون محیطی (EDTA) برای تست ژنتیکی
- در شرایط خاص، نمونه آمنیوتیک یا پرز کوریونی (برای بررسی پیش از تولد)
مقدار نمونه
- حداقل 3 تا ۵ میلیلیتر خون در لوله EDTA
- برای نمونه جنینی، ۱۰ میلیلیتر مایع آمنیوتیک یا نمونه CVS بنا بر سن بارداری
شرایط نگهداری نمونه
- نمونه خون باید در دمای ۲ تا ۸ درجه سانتیگراد نگهداری شود
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- در صورت استفاده از MLPA یا qPCR: حدود ۲ تا ۴ هفته
- در موارد ترکیبی با NGS یا تستهای خانوادگی: ۲ تا ۳ ماه
منابع علمی
- Sugarman EA, Nagan N, et al. “Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens.” European Journal of Human Genetics. 2012;20(1):27–32.
- Lefebvre S, Bürglen L, et al. “Identification and characterization of a spinal muscular atrophy–determining gene.” Cell. 1995;80(1):155–165.
- Prior TW, Krainer AR, Hua Y, et al. “Spinal muscular atrophy diagnostics: clinical and laboratory guidelines.” Muscle & Nerve. 2009;39(5):609–617.
- Darras BT, Monani UR, De Vivo DC. “Genetic Disorders Affecting the Motor Neuron: Spinal Muscular Atrophy.” Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Elsevier; 2015.
- Clinical Practice Guidelines for SMA, SMA Foundation & CureSMA. 2020.
")
")