درباره بیماری و روشهای تشخیص
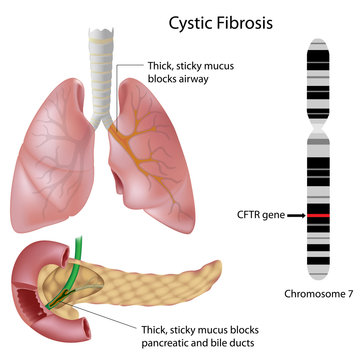
سیستیک فیبروزیس (CF) یک بیماری ژنتیکی و ارثی است که بر عملکرد غدد ترشحی بدن اثر میگذارد. این بیماری بهویژه ریهها و دستگاه گوارش را درگیر میکند. در بیماران مبتلا بهCF، ترشحات غلیظ و چسبناکی در بدن تولید میشود که باعث انسداد مجاری تنفسی، لوزالمعده و سایر اندامها میشود. شیوع این بیماری در کشورهای غربی بیشتر است. در اروپا و آمریکا، حدود ۱ در ۲۵۰۰ نوزاد به این بیماری مبتلا میشوند. در ایران شیوع پایینتری دارد، ولی بهعلت عدم غربالگری گسترده، بسیاری از موارد تشخیص داده نمیشوند.
عامل این بیماری، جهش در ژنCFTR (Cystic Fibrosis Transmembrane Conductance Regulator) است که در کروموزوم ۷ قرار دارد. این ژن مسئول تولید پروتئینی است که انتقال کلر و سدیم از غشاء سلولی را تنظیم میکند. اختلال در عملکرد این پروتئین، منجر به تولید ترشحات غیرطبیعی میشود.
علایم و نشانههای بالینی
علایم CF از دوران نوزادی شروع میشود و ممکن است شامل موارد زیر باشد:
- سرفه مزمن و تنفس دشوار
- عفونتهای مکرر تنفسی (مانند برونشیت و پنومونی)
- مشکلات گوارشی مانند مدفوع چرب، کاهش وزن و سوءجذب
- رشد ناکافی در دوران کودکی
- تعریق شور
- ناباروری در مردان ( به دلیل انسداد لوله های اسپرم)
روشهای تشخیص
- تست تعریق (Sweat Test): رایجترین روش؛ سطح کلر در تعریق بررسی میشود. در افراد مبتلا مقدار کلر بیش از ۶۰ میلیمول در لیتر است.
- آزمایش ژنتیک: بررسی جهش در ژنCFTR؛ در حال حاضر بیش از ۲۰۰۰ جهش شناسایی شده است، ولی جهش ΔF508 شایعترین نوع است.
- آزمایش آنزیمهای پانکراسی: برای ارزیابی عملکرد گوارشی
- آزمایش نوزادان (Newborn Screening): اندازهگیری سطح آنزیم IRT در خون خشک شده نوزادان
نوع نمونه
- خون محیطی
- تعریق (برای تست کلر)
- خون نوزاد خشک شده (برای غربالگری نوزادی)
مقدار نمونه
- خون محیطی: ۲ تا ۵ میلیلیتر
- نمونه تعریق: حداقل ۷۵ میلیگرم تعریق
- خون نوزاد (کاغذ گاتری): ۲ قطره بزرگ
شرایط نگهداری نمونه
- خون: در دمای ۲ تا ۸ درجه سانتیگراد تا ۷۲ ساعت
- تعریق: نمونه باید بلافاصله پس از جمعآوری تحلیل شود
- کاغذ گاتری: در دمای اتاق و دور از نور و رطوبت
مدت زمان جوابدهی
- تست تعریق: ۱ تا ۲ روز کاری
- آزمایش ژنتیک CFTR: 4 تا 6 هفته کاری
- غربالگری نوزادان: حداکثر ۷ روز پس از تولد
منابع علمی
- Cutting, G. R. (2015). “Cystic fibrosis genetics: from molecular understanding to clinical application.” Nature Reviews Genetics, 16(1), 45–56.
- Farrell, P. M., et al. (2017). “Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation.” The Journal of Pediatrics, 181, S4–S15.e1.
- Ratjen, F., & Döring, G. (2003). “Cystic fibrosis.” The Lancet, 361(9358), 681–689.
- O’Sullivan, B. P., & Freedman, S. D. (2009). “Cystic fibrosis.” The Lancet, 373(9678), 1891–1904.
- Elborn, J. S. (2016). “Cystic fibrosis.” The Lancet, 388(10059), 2519–2531.
")
")