درباره بیماری و روشهای تشخیص
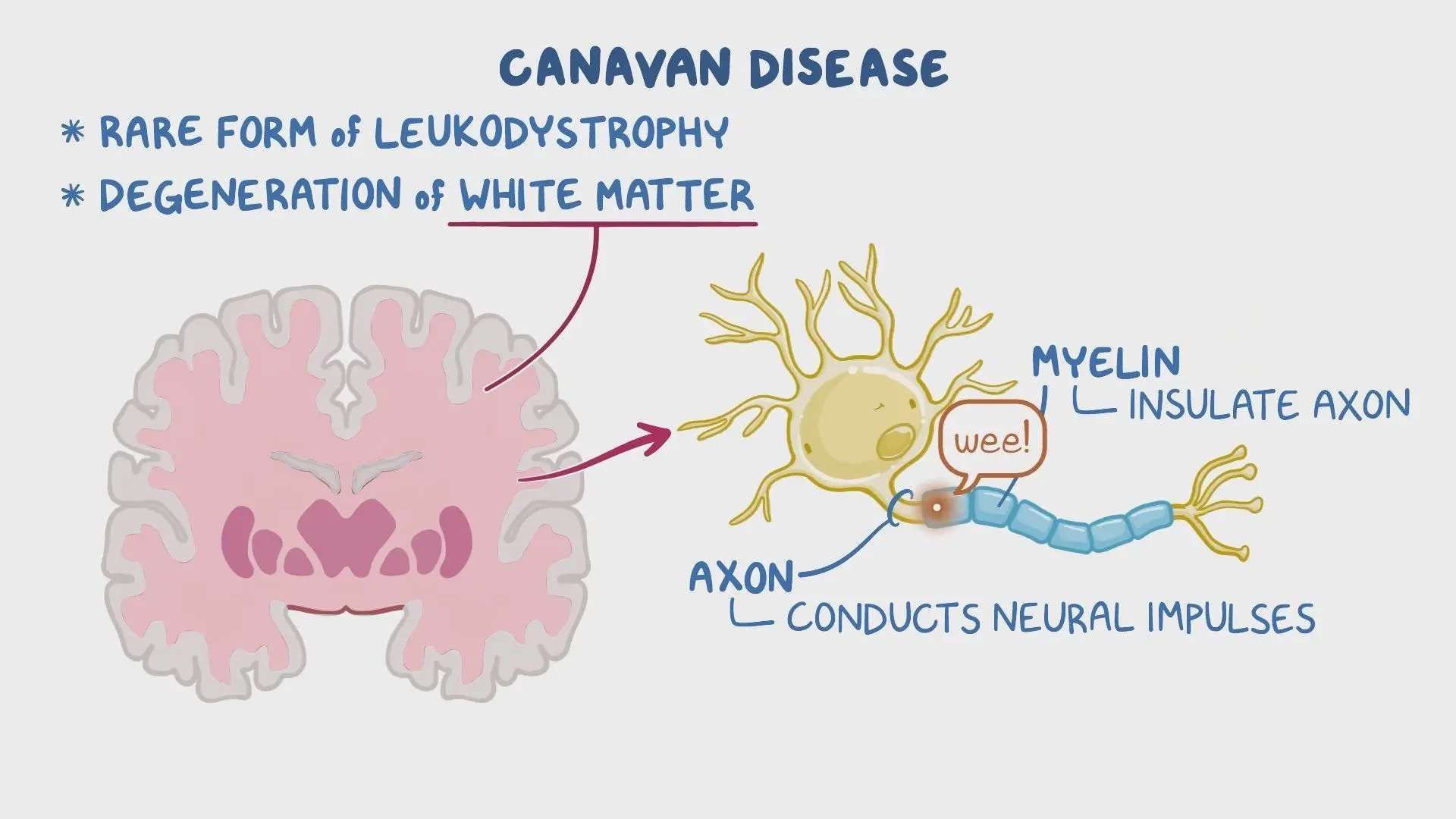
سندروم کاناوان یکی از بیماریهای نادر ولی شدید ژنتیکی در دستهی بیماریهای دژنراتیو سیستم عصبی مرکزی است که عمدتاً در دوران نوزادی یا اوایل کودکی بروز مییابد. این بیماری در نتیجهی جهش در ژن ASPA بهوجود میآید. این ژن مسئول تولید آنزیم آسپارتوآسیلاز است. این آنزیم نقش مهمی در تجزیهی مادهای به نام N -استیل آسپارتات (NAA) دارد. در نبود یا کاهش عملکرد این آنزیم، تجمع NAA در مغز باعث آسیب به میلین و تخریب بافتهای عصبی میشود.
انواع بیماری
دو نوع اصلی برای سندروم کاناوان شناسایی شده است:
- فرم نوزادی (کلاسیک): شدیدتر و زودرستر، با علایم واضح در شش ماه اول زندگی
- فرم نوجوانی: خفیفتر، با علایم دیررستر که ممکن است تا مدرسه رفتن تشخیص داده نشود.
علایم بیماری
علایم سندروم کاناوان معمولاً در سه تا شش ماهگی ظاهر میشود و با گذشت زمان شدت میگیرد. برخی از علایم رایج عبارت اند از:
- هیپوتونی (شل بودن عضلات)
- تاخیر در رشد حرکتی و ذهنی
- بزرگی غیرطبیعی سر (ماکروسفالی)
- اختلال در بلع، تنفس و تغذیه
- تشنج
- حرکات غیرطبیعی چشم (نیستاگموس)
متأسفانه بیشتر کودکان مبتلا به فرم کلاسیک این بیماری، تا سن بلوغ زنده نمیمانند.
تشخیص بیماری
تشخیص بیماری معمولاً از طریق روشهای زیر انجام میشود:
- آزمایش ادرار یا مایع مغزی- نخاعی (CSF): برای شناسایی سطح بالای NAA
- MRI مغز: نشاندهندهی دمیلینه شدن گسترده در ماده سفید مغز
- تست ژنتیکی: تأیید نهایی تشخیص با شناسایی جهش در ژن ASPA
در مواردی که سابقه خانوادگی وجود داشته باشد، غربالگری ژنتیکی قبل از تولد نیز توصیه میشود.
نوع نمونه
- خون محیطی
- ادرار یا مایع مغزی- نخاعی (CSF) برای اندازهگیری سطح NAA استفاده میشود.
مقدار نمونه
- خون محیطی: 3 تا 5 میلیلیتر
- ادرار: حداقل 10 میلیلیتر
- CSF: 1 تا 2 میلیلیتر
شرایط نگهداری نمونه
- خون محیطی: در دمای ۲ تا ۸ درجه سانتیگراد، ارسال در کمتر از ۷۲ ساعت
- ادرار و CSF: بلافاصله فریز شوند یا در دمای ۴ درجه نگهداری شوند و ظرف ۲۴ ساعت ارسال شوند.
مدت زمان جوابدهی
- تست ژنتیکی: بین 4 تا 6 هفته کاری
- تحلیل ادرار یا CSF: ۳ تا ۷ روز
منابع علمی
- Matalon, R., Michals, K., & Kaul, R. (1995). “Canavan disease: from spongy degeneration to molecular analysis.” Journal of Pediatrics, 127(4), 511–517.
- Kaul, R., Gao, G. P., & Matalon, R. (1994). “Canavan disease: mutations among Jewish and non-Jewish patients.” American Journal of Human Genetics, 55(1), 34–41.
- Adachi, M., Schneck, L., & Matalon, R. (1973). “Spongy degeneration of the brain in infancy (Canavan’s disease): clinical and pathologic study of 5 patients.” Neurology, 23(11), 1111–1122.
- Canavan Foundation. (2023). “Understanding Canavan Disease.” GeneReviews®. (2021). “Canavan Disease.” In: Adam MP, et al. (Eds.), University of Washington, Seattle.
")
")