درباره بیماری و روشهای تشخیص
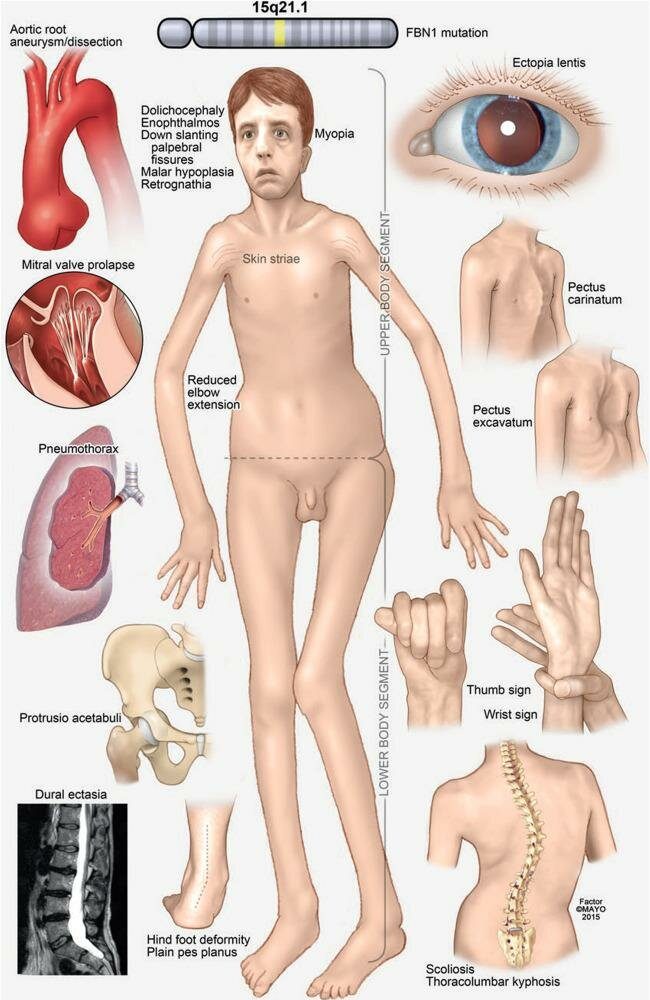
سندروم مارفان یک اختلال ژنتیکی نادر است که با درگیری بافت همبند بدن، بهویژه در قلب، رگهای خونی، چشمها، اسکلت و ریهها شناخته میشود. این سندروم بهصورت اتوزومال غالب به ارث میرسد و معمولاً به دلیل جهش در ژن FBN1 که کدکننده پروتئینی به نام فیبریلین-۱ است، ایجاد میگردد. فیبریلین-۱ نقش مهمی در استحکام و کشسانی بافت همبند دارد. علایم بیماری ممکن است در دوران کودکی یا نوجوانی ظاهر شود و با افزایش سن شدت یابد. اگرچه برخی افراد ممکن است تنها علایم خفیف اسکلتی داشته باشند، ولی خطر جدی معمولاً به دلیل آسیب آئورت و اختلالات قلبی- عروقی است که نیازمند پایش و درمان دقیق میباشند.
علایم و نشانههای بالینی
علایم سندروم مارفان از نظر شدت بسیار متغیر است، حتی در میان افراد یک خانواده. رایجترین ویژگیهای آن شامل:
- قد بلند با اندام کشیده و نازک
- دستها، بازوها، پاها و انگشتان بلندتر از حد طبیعی
- خمیدگی ستون فقرات (اسکولیوز) یا قوس شدید کف پا
- مشکلات چشمی مانند جابجایی عدسی چشم (ectopia lentis) و نزدیک بینی
- اتساع یا آنوریسم آئورت (که میتواند منجر به پارگی و مرگ شود)
- دریچههای قلبی غیرطبیعی، بهویژه پرولاپس دریچه میترال
تشخیص بیماری
تشخیص سندروم مارفان اغلب از طریق بررسی علایم بالینی و سابقه خانوادگی انجام میشود، ولی در بسیاری از موارد، تست ژنتیکی جهت شناسایی جهش در ژن FBN1 نیز انجام میگیرد.
همچنین از ابزارهای پاراکلینیکی زیر استفاده میشود:
- اکوکاردیوگرافی (ECHO) برای بررسی آئورت و دریچههای قلبی
- معاینه چشم و اسلیت لامپ
- تصویربرداری با CT یا MRI
نوع نمونه
- خون محیطی
- در کنار آن ممکن است تصویربرداریهای روتین قلب و عروق و معاینه چشم نیز انجام شود.
مقدار نمونه
- خون: 3 تا 5 میلیلیتر
شرایط نگهداری نمونه
- خون محیطی: در دمای ۲ تا ۸ درجه سانتیگراد، ارسال در کمتر از ۷۲ ساعت
مدت زمان جوابدهی
- آزمایش ژنتیک: 4 تا 6 هفته کاری
- سایر بررسیها مانند اکو یا معاینه چشم، در همان روز یا چند روز پس از انجام، آماده میشوند.
منابع علمی
- Dietz, H. C., Loeys, B., & De Backer, J. (2005). Marfan Syndrome. In: Adam MP, et al. (Eds.), GeneReviews® [Internet]. University of Washington, Seattle.
- Loeys, B. L., et al. (2010). “The revised Ghent nosology for the Marfan syndrome.” Journal of Medical Genetics, 47(7), 476-485.
- Pyeritz, R. E. (2014). “Marfan syndrome: improved clinical history results in expanded natural history.” Genetics in Medicine, 16(10), 717–724.
- Faivre, L., Collod-Beroud, G., et al. (2008). “Pathophysiological mechanism and clinical phenotype of Marfan syndrome: mutation analysis of FBN1.” Human Mutation, 29(8), 958–968.
- National Marfan Foundation. (2024). “Understanding Marfan Syndrome.”
")
")