درباره بیماری و روشهای تشخیص
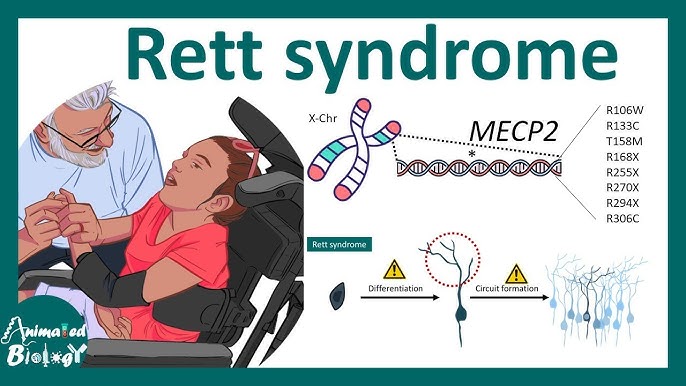
سندروم رِت یک بیماری عصبی- رشدی نادر است که بیشتر در دختران دیده میشود و روند رشد طبیعی کودک را پس از ماههای ابتدایی زندگی مختل میسازد. این اختلال معمولاً تا ۶ تا ۱۸ ماهگی علایمی ندارد، اما پس از آن، کاهش مهارتهای حرکتی، زبانی و شناختی بهتدریج آغاز میشود. سندروم رت بهعنوان یکی از مهمترین علل عقبماندگی ذهنی پیشرونده در دختران شناخته میشود.
سندروم رِت اغلب به دلیل جهش در ژن MECP2 واقع بر کروموزوم X ایجاد میشود. این ژن در تنظیم بیان سایر ژنها در مغز نقش مهمی دارد. در پسران، این جهش معمولاً کشنده است، چرا که کروموزوم X دوم ندارند. بیشتر موارد بیماری به صورت اسپورادیک (جهش جدید در کودک) رخ میدهد و ارثی نیست.
مراحل بالینی
- مرحله اول (کند شدن رشد): کاهش علاقه به محیط، تأخیر در مهارتهای حرکتی
- مرحله دوم (تخریب سریع): از دست دادن مهارتهای زبانی، حرکات هدفمند دست، بروز حرکات تکراری مانند مالیدن دستها
- مرحله سوم (ایست نسبی): بهبودی نسبی در برخی عملکردها، اما اختلالات حرکتی ادامهدار
- مرحله چهارم (زوال حرکتی): مشکلات شدید حرکتی، سفتی عضلات، اسکولیوز و مشکلات بلع
علایم و نشانههای بالینی
- از بین رفتن توانایی صحبت کردن و حرکات هدفمند دست
- حرکات تکراری و کلیشهای دستها مانند مالیدن، شستن یا فشار دادن
- کاهش رشد محیط سر (میکروسفالی اکتسابی)
- صرع در بسیاری از موارد
- اختلال در تعادل، راه رفتن و هماهنگی عضلات
- ناتوانی ذهنی متوسط تا شدید
- مشکلات تغذیه، تنفس و خواب
- در برخی موارد، ویژگیهای اوتیسم مانند در اوایل بیماری دیده میشود.
روشهای تشخیص
- تشخیص بالینی بر اساس روند رشد و علایم کلاسیک
- بررسی ژنتیکی مولکولی برای ژن MECP2 در بیش از ۹۵٪ موارد کلاسیک جهش شناسایی میشود.
- نوار مغزی (EEG): برای بررسی تشنجها
- گاهی برای افتراق از اختلالات مشابه مانند اوتیسم، آزمایشهای تکمیلی دیگر انجام میشود.
نوع نمونه
- خون محیطی
- مایع آمنیوتیک در تشخیص پیش از تولد
مقدار نمونه
- خون: ۳ تا ۵ میلیلیتر
- مایع آمنیوتیک: 20 میلی لیتر
شرایط نگهداری نمونه
- خون محیطی: در دمای ۲ تا ۸ درجه سانتیگراد، ارسال در کمتر از ۷۲ ساعت
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- آزمایش مولکولی برای ژنMECP2 : حدود 4 تا 6 هفته کاری
- در صورت تشخیص پیش از تولد (PND): حدود 4 تا 6 هفته کاری
منابع علمی
- Neul, J. L., et al. (2010). “Rett syndrome: revised diagnostic criteria and nomenclature.” Annals of Neurology, 68(6), 944–950.
- Amir, R. E., et al. (1999). “Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.” Nature Genetics, 23(2), 185–188.
- Percy, A. K. (2016). “Rett syndrome: new perspectives and clinical challenges.” Neurology, 86(22), 1986–1991.
- Archer, H., et al. (2006). “MECP2 mutations: new insights into the pathogenesis of Rett syndrome.” Journal of Medical Genetics, 43(5), 377–384.
- National Institute of Neurological Disorders and Stroke (NINDS). (2023). “Rett Syndrome Information Page.”
")
")