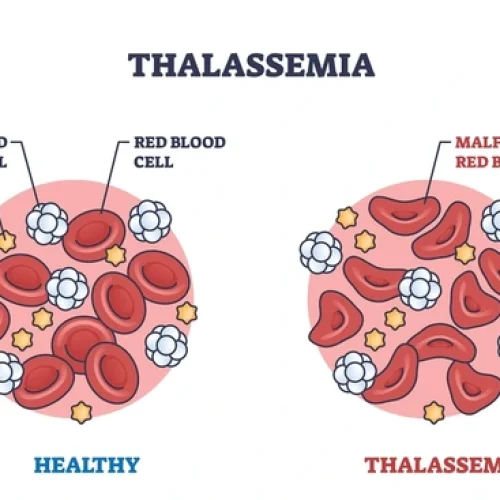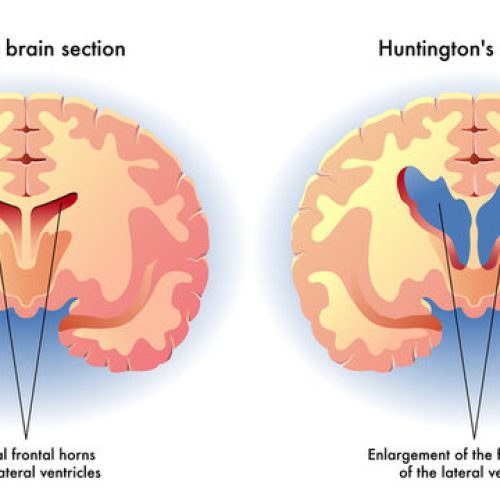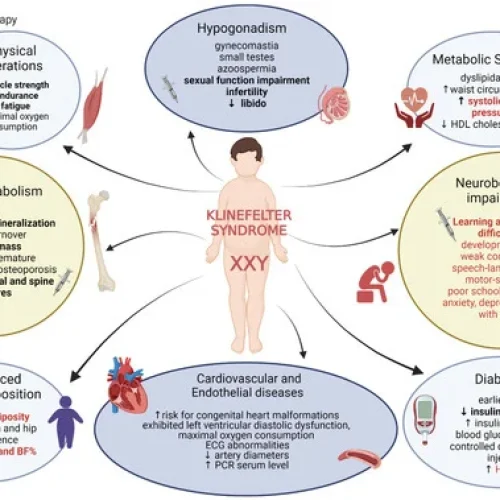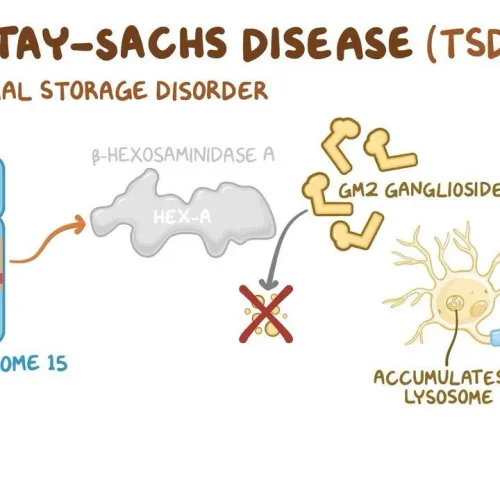
درباره بیماری و روشهای تشخیص
سندروم دیجورج که با نام علمی حذف22q11.2 نیز شناخته میشود، یک اختلال ژنتیکی است که ناشی از حذف بخش کوچکی از کروموزوم ۲۲ در موقعیت q11.2 است. این سندروم طیف وسیعی از علایم و مشکلات جسمی، ایمنی، قلبی، و رشدی را ایجاد میکند. این بیماری یکی از شایعترین سندرومهای ناشی از حذف کروموزومی است و تقریباً در هر ۳۰۰۰ تا ۶۰۰۰ تولد یک مورد از آن دیده میشود.
در اکثر بیماران، سندروم دیجورج ناشی از حذف میکروسکوپی بخشی از بازوی بلند کروموزوم 22 است. این حذف میتواند به صورت خودبهخود (de novo) یا ارثی باشد. حدود ۹۰٪ موارد به طور خود به خود رخ میدهد و ۱۰٪ دیگر به صورت وراثت اتوزومال غالب از یکی از والدین منتقل میشود.
علایم و نشانههای بالینی
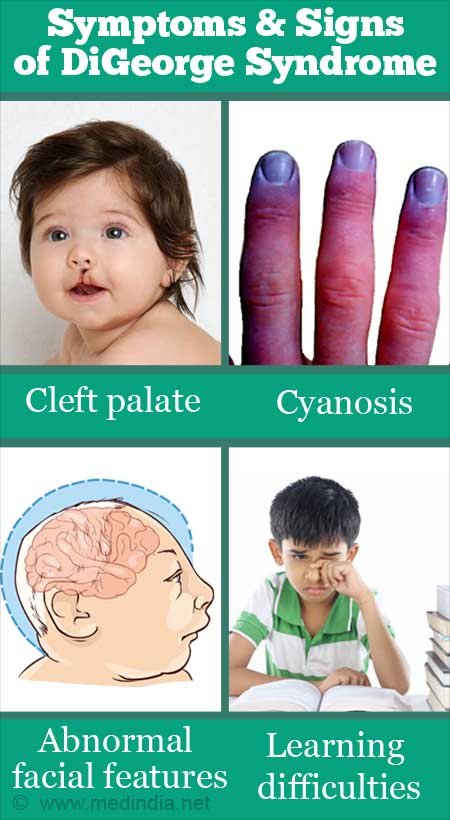
- نقایص قلبی مادرزادی (مانند تترالوژی فالوت، ترانپوزیشن عروق بزرگ، نقص تیغه بینبطنی)
- نقص در سیستم ایمنی (به دلیل هیپوپلازی یا فقدان تیموس)
- نقایص کام و لب (شکاف کام نرم یا کام ناقص)
- مشکلات یادگیری، گفتاری و روانی
- کاهش سطح کلسیم خون (به دلیل هیپوپاراتیروئیدیسم)
- اختلالات رفتاری یا روانپزشکی (مانند اختلال نقص توجه، اضطراب، شیزوفرنی)
روشهای تشخیص
- آزمایش ژنتیکی مولکولی FISH یا MLPA یا Microarray برای شناسایی حذف 22q11.2
- اکوکاردیوگرافی: بررسی ناهنجاریهای قلبی
- آزمایش سطح کلسیم و عملکرد پاراتیروئید
- بررسی عملکرد سیستم ایمنی (شمارش سلولهای T)
- ارزیابی روانی و یادگیری، گفتاردرمانی
تشخیص قطعی از طریق آزمایش ژنتیکی انجام میشود و در کودکان با نقص قلبی یا شکاف کام، توصیه به بررسی این سندروم میشود.
نوع نمونه
- خون محیطی
- مایع آمنیوتیک یا نمونه CVS برای بررسی پیش از تولد
مقدار نمونه
- خون: ۳ تا ۵ میلیلیتر
- مایع آمنیوتیک: 20 میلیلیتر
شرایط نگهداری نمونه
- خون: ۲ تا ۸ درجه سانتیگراد تا ۷۲ ساعت
- مایع آمنیوتیک : دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- تست FISHیا MLPA: ۷ تا ۱۰ روز کاری
- تست Array-CGH : حدود 4 تا 6 هفته
- تشخیص پیش از تولد: حدود 4 تا 6 هفته
منابع علمی
- McDonald-McGinn, D. M., et al. (2015). “22q11.2 deletion syndrome.” Nature Reviews Disease Primers, 1, 15071.
- Sullivan, K. E. (2008). “Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome.” Immunology and Allergy Clinics of North America, 28(2), 353–366.
- Ryan, A. K., et al. (1997). “Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study.” Journal of Medical Genetics, 34(10), 798–804.
- Devriendt, K., et al. (1998). “DiGeorge syndrome: 22q11 deletion syndrome.” Lancet, 351(9119), 1101–1105.
- Digilio, M. C., et al. (2003). “Clinical features of the 22q11 deletion syndrome: a multicenter study.” American Journal of Medical Genetics Part A, 123A(2), 127–133.
")
")