درباره بیماری و روشهای تشخیص
سندروم آنجلمن یک اختلال ژنتیکی نادر است که با تأخیر شدید در رشد، اختلال در گفتار، مشکلات تعادل و حرکت، و رفتارهای شاد و خندهرو همراه است. این سندروم در ابتدا در سال 1965 توسط پزشک انگلیسی «هری آنجلمن» توصیف شد و به افتخار او نامگذاری شده است. این بیماری تقریباً در ۱ نفر از هر ۱۵۰۰۰ تا ۲۰۰۰۰ تولد رخ میدهد.
سندروم آنجلمن معمولاً به دلیل نقص در بیان ژن UBE3A در کروموزوم 15 در ناحیه 15q11-q13 ایجاد میشود. این ژن در مغز عمدتاً فقط از نسخه مادری فعال است و نسخه پدری خاموش میماند. هرگونه اختلال در نسخه مادری مانند:
- حذف کروموزومی مادری (deletion)
- موتاسیون (جهش) در ژن UBE3A
- عدم دریافت ژن مادری به دلیل پدیدهای به نام دایزومی تکوالدی (Uniparental disomy)
- اختلال در مرکز کنترل ایمپرینتینگ (IC)
میتواند منجر به بروز سندروم آنجلمن شود.
علایم و نشانههای بالینی
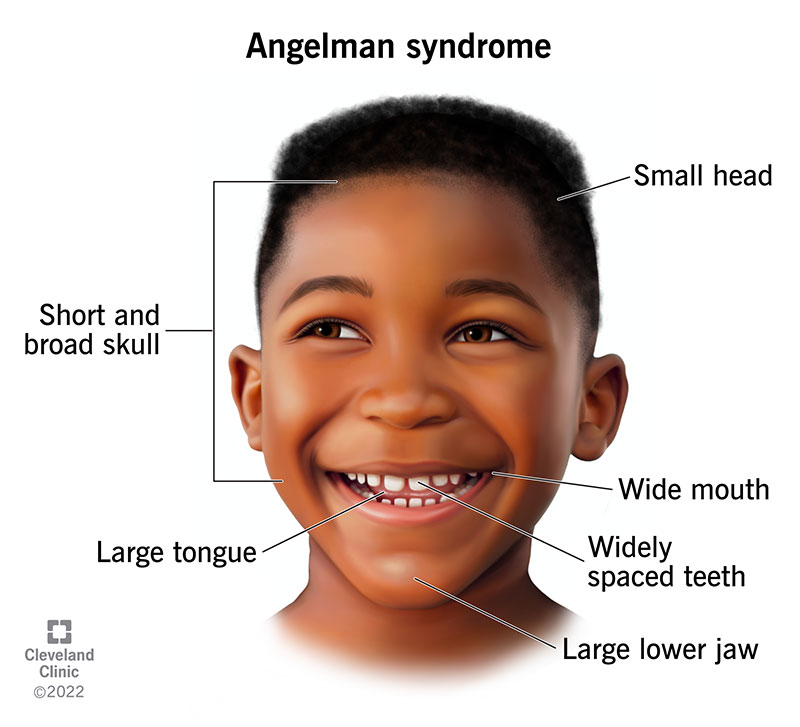
ویژگیهای بالینی سندروم آنجلمن بهتدریج در سال اول زندگی ظاهر میشوند و شامل موارد زیر هستند:
- تأخیر شدید در تکامل روانی و حرکتی
- عدم گفتار یا گفتار بسیار محدود
- حرکات نامتناسب و آتاکسیک (مشکل در تعادل و هماهنگی عضلات)
- رفتار شاد، خندههای مکرر، تحریکپذیری، و علاقه به آب
- تشنج (در بیش از 80% بیماران)
- مشکلات خواب، بیشفعالی و بیقراری
- ویژگیهای چهرهای خاص مانند دهان باز، زبان بیرونزده، لبخندهای مکرر
روشهای تشخیص
تشخیص سندروم آنجلمن معمولاً بر اساس علایم بالینی و سپس تستهای ژنتیکی انجام میشود:
- مطالعه متیلاسیون DNA (MS-PCR): بررسی متیلاسیون در ناحیه 15q11-q13 رای تشخیص حذف، ایمپرینتینگ یا دایزومی تکوالدی.
- تست Array-CGH: برای شناسایی حذف کروموزومی
- توالییابی ژن UBE3A: برای یافتن جهش در صورت منفی بودن تستهای قبلی
- آزمایشهای الکتروانسفالوگرافی (EEG): برای بررسی الگوی خاص امواج مغزی که در این سندروم دیده میشود.
- تصویربرداری مغزی (MRI): معمولاً طبیعی است اما در مواردی ناهنجاریهای خفیف مشاهده میشود.
نوع نمونه
- خون محیطی
- مایع آمنیوتیک یا CVS برای تشخیص پیش از تولد
مقدار نمونه
- خون محیطی: ۳ تا ۵ میلیلیتر
- مایع آمنیوتیک: 20 میلیلیتر
شرایط نگهداری نمونه
- خون محیطی: ۲ تا ۸ درجه سانتیگراد تا ۷۲ ساعت
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- تست متیلاسیون DNA: حدود ۱۰ تا ۱۴ روز کاری
- تست Array-CGH: حدود 4 تا 6 هفته
- توالییابی ژن UBE3A: ۴ تا ۶ هفته
- تشخیص پیش از تولد: حدود 4 تا 6 هفته
منابع علمی
- Williams, C. A., et al. (2006). “Angelman syndrome: consensus for diagnostic criteria.” American Journal of Medical Genetics Part A, 140A(5), 413–418.
- Margolis, S. S., et al. (2015). “UBE3A, a gene implicated in Angelman syndrome, regulates synapse development and maintenance.” Nature Neuroscience, 18(5), 683–689.
- Dagli, A. I., et al. (2012). “Angelman syndrome.” GeneReviews® [Internet]. University of Washington, Seattle.
- Lossie, A. C., et al. (2001). “Distinct phenotypes distinguish the molecular classes of Angelman syndrome.” Journal of Medical Genetics, 38(12), 834–845.
- Tan, W. H., et al. (2011). “Angelman syndrome: mutations influence features in early childhood.” American Journal of Medical Genetics Part A, 155(1), 81–90.
")
")