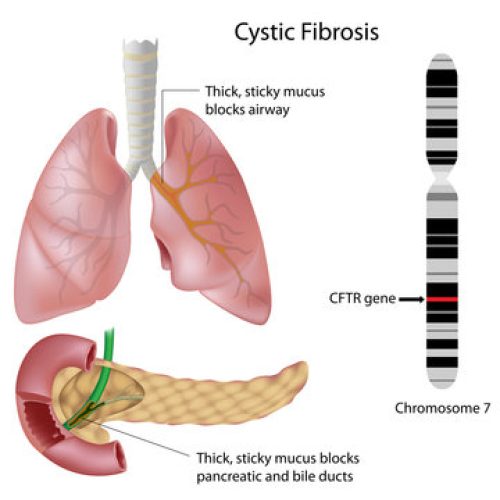
درباره بیماری و روشهای تشخیص
رتینیت پیگمنتوزا یک گروه از بیماریهای ژنتیکی نادر چشم است که باعث تخریب تدریجی سلولهای شبکیه، به ویژه سلولهای گیرنده نور (استوانهای و مخروطی)، میشود. شبکیه، لایهای نازک در پشت چشم است که نور را دریافت و آن را به سیگنالهای عصبی تبدیل میکند. در بیماران مبتلا به RP، این سلولها بهتدریج میمیرند، که منجر به کاهش بینایی پیشرونده و در موارد شدید کوری کامل میشود.
الگوهای وراثتی
رتینیت پیگمنتوزا میتواند به شکلهای مختلفی به ارث برسد:
- وراثت اتوزومال مغلوب: شایعترین نوع، که در صورت وجود دو نسخه معیوب ژن بروز مییابد.
- وراثت اتوزومال غالب: تنها یک نسخه معیوب از ژن برای بروز بیماری کافی است.
- وراثت وابسته به کروموزوم X: معمولاً مردان را مبتلا میکند، در حالی که زنان ناقل بیماری هستند.
- در برخی موارد، بیماری به صورت جهش جدید (de novo) رخ میدهد.
ژنهای مرتبط
بیش از 100 ژن با RP در ارتباط هستند، از جمله:
- RPGR برای فرمهای وابسته به X
- RHO برای فرمهای اتوزومال غالب
- USH2A، EYS، CRB1 و PRPH2
برخی از این ژنها تنها در گیرنده های نوری شبکیه فعال هستند.
علایم و نشانههای بالینی
علایم RP معمولاً از دوران نوجوانی یا جوانی آغاز میشود:
- شبکوری (Nyctalopia): کاهش دید در محیطهای تاریک
- دید تونلی: محدود شدن میدان دید محیطی
- کاهش دید مرکزی در مراحل پیشرفته
- حساسیت به نور و تاری دید
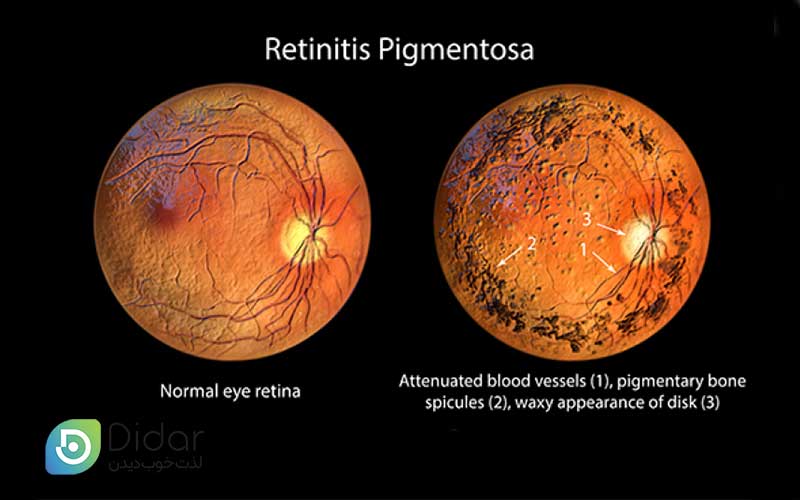
- در معاینه چشمپزشکی: لکههای تیره رنگدانهای مشخص در شبکیه (ظاهر استخوان ماهیچهای)
روشهای تشخیص
- معاینه چشم کامل: توسط چشمپزشک با افتالموسکوپ
- الکترو رتینوگرام (ERG): بررسی عملکرد گیرندههای نوری
- تصویربرداریOCT: مشاهده ساختار شبکیه
- تست میدان بینایی (Perimetry): تعیین وسعت دید
- آزمایش ژنتیک: انجام NGS برای تعیین ژن معیوب و نوع وراثت
نوع نمونه
- نمونه خون محیطی
- بررسیهای چشمی با دستگاههای تخصصی نیازی به نمونهگیری ندارند.
مقدار نمونه
- خون محیطی: ۲ تا ۵ میلیلیتر
شرایط نگهداری نمونه
- خون: در دمای ۲ تا ۸ درجه سانتیگراد تا ۷۲ ساعت
مدت زمان جوابدهی
- معاینه چشم پزشکی و ERG: در همان روز یا حداکثر ۱ تا ۲ روز
- نتایج OCT و تصویربرداری شبکیه: همان روز
- تست های ژنتیک: 2 تا 3 ماه
منابع علمی
- Daiger, S. P., Bowne, S. J., & Sullivan, L. S. (2014). “Genes and mutations causing retinitis pigmentosa.” Clinical Genetics, 84(2), 132-141.
- Hartong, D. T., Berson, E. L., & Dryja, T. P. (2006). “Retinitis pigmentosa.” The Lancet, 368(9549), 1795-1809.
- Hamel, C. (2007). “Retinitis pigmentosa.” Orphanet Journal of Rare Diseases, 2(1), 40.
- den Hollander, A. I., Black, A., Bennett, J., & Cremers, F. P. M. (2010). “Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies.” Journal of Clinical Investigation, 120(9), 3042-3053.
- Fahim, A. T., Daiger, S. P., & Weleber, R. G. (2017). “Retinitis pigmentosa overview.” GeneReviews®, University of Washington, Seattle.
")
")