درباره بیماری و روشهای تشخیص
آلکاپتونوریا یک بیماری متابولیک نادر و ارثی است که در اثر نقص در تجزیه یک اسید آمینه به نام تیروزین به وجود میآید. علت این بیماری، جهش در ژن HGD (Homogentisate 1,2-dioxygenase) است که موجب کمبود آنزیمی به همین نام میشود. در نتیجه، مادهای به نام هوموجنتیسیک اسید (HGA) در بدن انباشته شده و با گذر زمان به بافتها و استخوانها آسیب میزند.
این بیماری از دستهی اختلالات اتوزومی مغلوب است؛ یعنی فرد برای بروز بیماری باید هر دو نسخهی ژن معیوب را از پدر و مادر به ارث ببرد.
علایم و نشانههای بالینی
- ادرار تیرهرنگ: در اثر اکسید شدنHGA ، ادرار در تماس با هوا به رنگ قهوهای یا سیاه درمیآید (یکی از اولین نشانهها در دوران نوزادی)
- آلکاپتونوریایی: اصطلاحی که به وجود HGA در ادرار اشاره دارد.
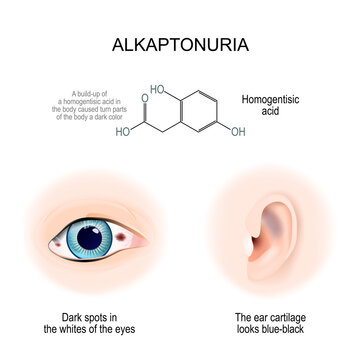
- اُکرونوز(Ochronosis) : رسوب رنگدانههای تیره در غضروفها، پوست، صلبیه چشم و مفاصل
- درد و تخریب مفاصل: خصوصاً در ستون فقرات و زانوها (از دهه سوم یا چهارم زندگی)
- سنگ کلیه یا پروستات
- در موارد پیشرفته، ممکن است قلب، دریچههای قلبی یا کلیهها درگیر شوند.
روشهای تشخیص
- آزمایش ادرار: شناسایی HGA با روشهای شیمیایی یا کروماتوگرافی
- تست Ferric chloride یا Benedict: واکنش رنگی مثبت در ادرار
- تست ژنتیکی: شناسایی جهش در ژن HGD برای تأیید تشخیص
- تصویربرداری مفاصل: در صورت درگیری اسکلتی جهت ارزیابی تخریب مفاصل
تشخیص زودهنگام و بررسی سابقه خانوادگی در پیشگیری از آسیبهای جدی در سنین بالاتر اهمیت دارد.
نوع نمونه
- ادرار تازه برای سنجش سطح HGA
- خون محیطی
- مایع آمنیوتیک در تشخیص پیش از تولد
مقدار نمونه
- ادرار: حداقل ۵ تا ۱۰ میلیلیتر
- خون: ۳ تا ۵ میلیلیتر
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
شرایط نگهداری نمونه
- ادرار: در ظرف تمیز، بدون نگهدارنده، در دمای یخچال؛ حداکثر تا ۲۴ ساعت
- خون: در دمای ۲ تا ۸ درجه سانتیگراد؛ ارسال طی ۲ تا ۳ روز
- مایع آمنیوتیک: دمای ۲۰ تا ۲۳ درجه و ارسال سریع
مدت زمان جوابدهی
- آنالیز ادرار: ۲ تا ۵ روز کاری
- آزمایش ژنتیک: 4 تا 6 هفته
- پاسخ بررسی پیش از تولد (در صورت شناخته بودن جهش): ۱۰ تا ۱۴ روز کاری
منابع علمی
- Phornphutkul, C., Introne, W. J., Perry, M. B., et al. (2002). “Natural history of alkaptonuria.” New England Journal of Medicine, 347(26), 2111–2121.
- Ranganath, L. R., et al. (2011). “Alkaptonuria: a review of surgical and non-surgical clinical aspects.” Orphanet Journal of Rare Diseases, 6(1), 82.
- Zatkova, A., et al. (2012). “Alkaptonuria: current perspectives.” APMIS, 120(6), 400–406.
- Khedr, M., et al. (2020). “New insights into the pathophysiology and management of alkaptonuria.” Clinical Genetics, 98(4), 293–301.
- National Organization for Rare Disorders (NORD). (2022). “Alkaptonuria.” Available at:
")
")